Datorķīmija: kas tā ir — metodes, īpašības un pielietojums
Datorķīmija: uzzini, kas tā ir, metodes, īpašības un pielietojums — molekulu modelēšana, zāļu un materiālu izstrāde, reakciju prognozes un spektroskopija.
Datorķīmija ir ķīmijas nozare, kurā ķīmijas problēmu risināšanai izmanto datorzinātnes. Šīs programmas aprēķina molekulu un cietu vielu struktūras un īpašības. Datorķīmija parasti papildina informāciju, kas iegūta ar ķīmiskiem eksperimentiem. Tā var paredzēt ķīmiskas parādības, kas vēl nav novērotas. To plaši izmanto jaunu zāļu un materiālu izstrādē.
Datorķīmija var paredzēt struktūru (t. i., paredzamo molekulas atomu atrašanās vietu), absolūto un relatīvo (mijiedarbības) enerģiju, elektroniskā lādiņa sadalījumu, dipolus un augstākos daudzpolu momentus, vibrācijas frekvences, reaktivitāti vai citus spektroskopiskos lielumus, kā arī sadursmju ar citām daļiņām šķērsgriezumus.
Datorķīmija aplūko gan statiskas, gan dinamiskas sistēmas. Visos gadījumos, palielinoties pētāmās sistēmas lielumam, pieaug arī izmantotais datora laiks un citi resursi (piemēram, atmiņa un vieta diskā). Šī sistēma var būt viena molekula, molekulu grupa vai cieta viela. Datorķīmijas metožu diapazons ir no ļoti precīzām līdz ļoti aptuvenām. Ļoti precīzas metodes parasti ir izmantojamas tikai mazām sistēmām.
Attēlu galerija
8 Attēli


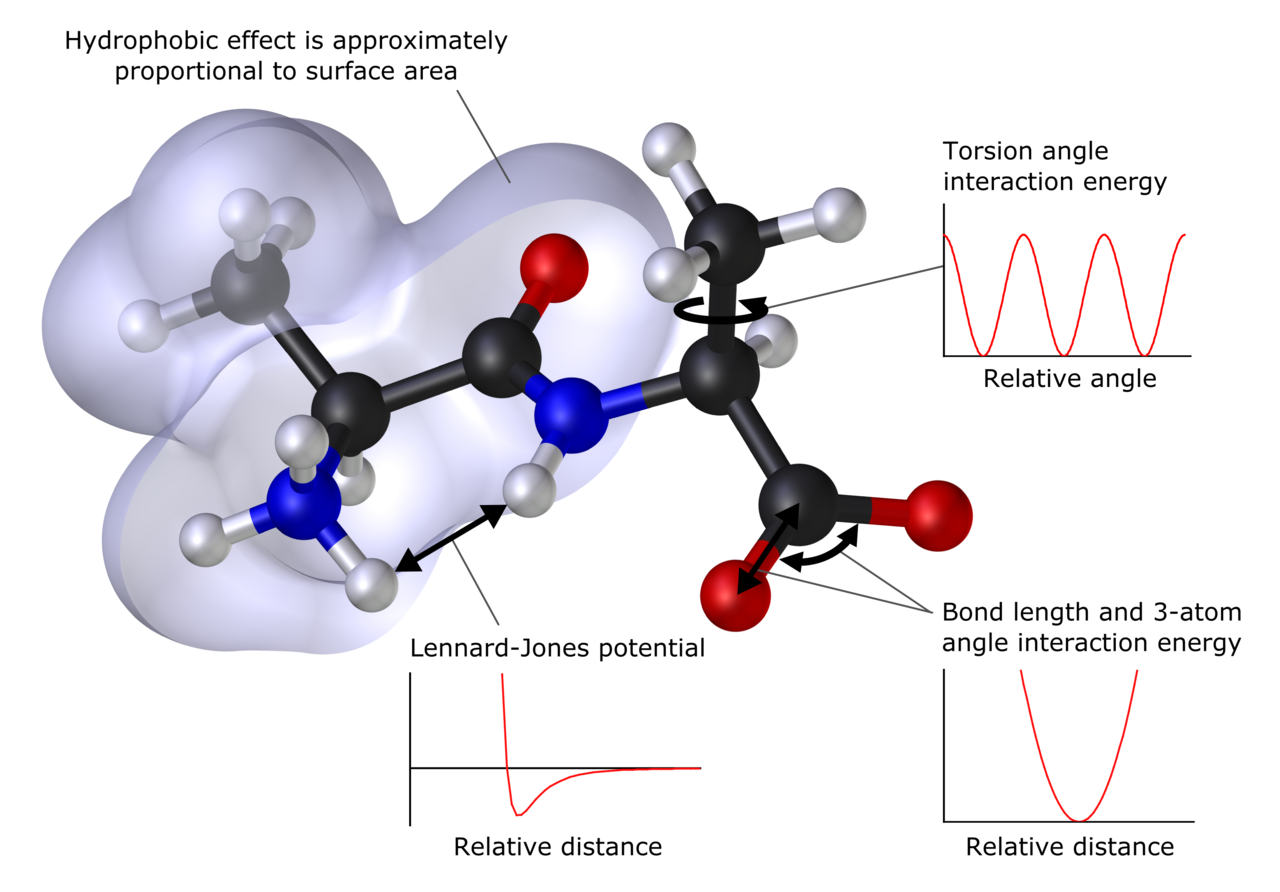


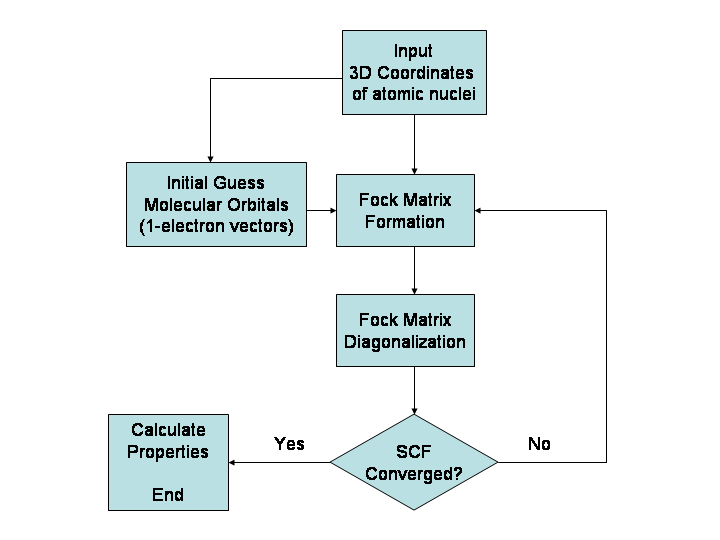
Galvenās metodes
- Ab initio (no pamatprincipiem) — kvantķīmiskas metodes, piemēram, Hartree–Fock (HF) un post-HF metodes (MP2, CCSD(T) u. c.), kuras pamatojas uz Schrēdingera vienādojumu. Ļauj iegūt ļoti precīzus rezultātus, taču strauji aug aprēķinu izmaksas ar sistēmas izmēru.
- Blīvuma funkciju teorija (DFT) — plaši izmantota kvantķīmijas pieeja, kas bieži nodrošina labu precizitātes/izmaksu attiecību molekulu un cieto vielu aprēķinos. Rezultātu kvalitāte atkarīga no izvēlētā funkciona.
- Semiempīriskās metodes — vienkāršotas kvantķīmiskas metodes, kurās daļa parametrizācijas balstīta uz eksperimentiem. Ātrākas, bet mazāk precīzas nekā „no pamatprincipiem” metodes.
- Molekulārā mehānika (MM) — klasiskas daļiņu mijiedarbības apraksti, kas izmanto potenciāla laukus (spēku laukumus). Ļoti ātras un piemērotas lielām sistēmām (biomolekulām, polimēriem), bet neparedz elektroniskās izmaiņas ķīmiskās reakcijās.
- Molekulārā dinamika (MD) — laika atkarīgu procesu simulācijas, kurās aprēķina atomu kustības, izmantojot MM vai reizēm QM aprakstus. Izmanto, lai pētītu termiskās īpašības, konformācijas un kinētiku.
- Hibrīdmetodes (QM/MM) — apvieno kvantķīmiju reaktīvā centra aprakstam (QM) ar molekulārās mehānikas modeli apkārtējai videi (MM). Bieži lieto biokatalīzes un enzīmu reakciju pētīšanai.
Kādus lielumus un īpašības var paredzēt
- Struktūras: atomu ģeometrijas, sākot no vienkāršiem savienojumiem līdz kristālu režģiem.
- Enerģijas: absolūtās enerģijas, reakcijas enerģijas, aktivācijas barjeras.
- Elektroniskās īpašības: lādiņu sadalījums, orbitales, joslu struktūras cietvielām.
- Spektroskopiskie lielumi: IR/Raman vibrācijas, NMR ķīmiskie posmi, UV‑Vis pārejas, EPR parametri.
- Termodinamikas un kinētikas raksturojumi: brīvās enerģijas aprēķini, reakciju ceļu identifikācija, pārejas stāvokļu meklēšana.
- Interakcijas un saķeres: molekulu savstarpējās mijiedarbības, saistošās brīvības, sadursmju šķērsgriezumi.
Pielietojumi
- Zāļu izstrāde un molekulārā dizaina atbalsts — molekulāra sajūga prognozēšana (docking), farmakoforu veidošana, ADMET prognozes un apmeklēšana pirms eksperimentu veikšanas.
- Jaunu materiālu izstrāde — pusvadītāji, katalizatori, bateriju materiāli, polimēri ar īpašām mehāniskajām vai optiskajām īpašībām.
- Katalīzes un reakciju mehāniku izpēte — aktivācijas barjeru aprēķins, katalītisko ceļu optimizācija.
- Spektroskopisko datu interpretācija — palīdz skaidrot eksperimentālus spektrus un identificēt struktūras.
- Vides ķīmija un toksikoloģija — reakciju ceļu prognozēšana, piesārņojuma sadalīšanās, toksicitātes modeļi.
- Biomolekulu dinamika — proteīnu kustības, ligand–receptors mijiedarbības, membrānu uzvedība.
Ierobežojumi un grūtības
- Skalējamība: precīzākās metodes parasti ir piesaistītas ļoti maziem sistēmas izmēriem — aprēķinu laiks un atmiņas prasības strauji pieaug.
- Modeļu aproksimācijas: rezultāti atkarīgi no izvēlētā metodes līmeņa, funkciona, bāzes kopas vai spēka lauka parametrizācijas.
- Temperatūras un vidi ietekme: daudzi aprēķini tiek veikti vakuumā vai ar vienkāršotu vidi; solvatācijas efekti un entropijas kontribūcijas prasa papildu aprēķinus.
- Validācija: teorētiskie rezultāti jāsalīdzina ar eksperimentiem, lai novērtētu ticamību.
Programmatūra un aparatūra
Bieži izmantotās programmas: Gaussian, ORCA, VASP, CP2K, GAMESS, NWChem, GROMACS, AMBER, CHARMM un citas. Izvēle atkarīga no vajadzīgās metodes un sistēmas tipa. Datorķīmiķi izmanto gan klasterus un superdatorus, gan GPU paātrinātas sistēmas un mākoņresursus, lai samazinātu aprēķinu laiku.
Tipiska darba gaita
- Definēt problēmu un izvēlēties piemērotu metožu līmeni.
- Sagatavot sākotnējo struktūru un parametrus (bāzes kopas, funkcioni, spēka lauki).
- Veikt optimizācijas un nepieciešamos dinamiskos aprēķinus.
- Aprēķināt īpašības (spektroskopiju, enerģijas, pārejas stāvokļus).
- Salīdzināt ar eksperimentiem un iterēt modeli, ja nepieciešams.
Nākotnes virzieni
Strauji attīstās mašīnmācīšanās metodes — ML potenciāli un ģeneratīvi modeļi, kas paātrina enerģiju un spēku laukus aprēķinus ar eksperimentālu vai augstas precizitātes datu palīdzību. Arī kvantuaprēķini sola jaunas iespējas kvantķīmijas problēmu risināšanā, lai gan praktiskai pielietošanai vēl nepieciešama tehnoloģiju nobriešana.
Praktiski padomi iesācējiem
- Sāciet ar vienkāršiem piemēriem un pakāpeniski palieliniet metodes sarežģītību.
- Pārbaudiet rezultātus ar dažādām metodēm un bāzes kopām, lai novērtētu stabilitāti.
- Vienmēr salīdziniet teorētiskos rezultātus ar eksperimentiem, ja tie ir pieejami.
- Dokumentējiet parametrus un iestatījumus reproducējamībai.
Datorķīmija ir spēcīgs instruments, kas savieno teoriju, skaitļošanu un eksperimentu, ļaujot paredzēt un izprast ķīmiskās sistēmas dziļākā līmenī. Ar pareizu metožu izvēli un rūpīgu validāciju tā būtiski paātrina jaunu materiālu un zāļu attīstību, samazinot izmaksas un eksperimentālo darbu.

Saistītās lapas
- Bioinformātika
- Statistiskā mehānika
Jautājumi un atbildes
J: Kas ir skaitļošanas ķīmija?
A: Datorķīmija ir ķīmijas nozare, kurā izmanto datorzinātnes, lai palīdzētu risināt ķīmijas problēmas. To var izmantot, lai aprēķinātu molekulu un cietu vielu struktūras un īpašības, prognozētu ķīmiskas parādības, kas vēl nav novērotas, un izstrādātu jaunas zāles un materiālus.
J: Kāda veida sistēmas aplūko skaitļošanas ķīmija?
A: Datorķīmija aplūko gan statiskas, gan dinamiskas sistēmas. Sistēma var būt viena molekula, molekulu grupa vai cieta viela.
J: Kāda veida informāciju var sniegt skaitļošanas ķīmija?
A: Datorķīmija var sniegt tādu informāciju kā struktūra (atomu atrašanās vietas), absolūtā un relatīvā enerģija, elektroniskā lādiņa sadalījums, dipoli un augstāko multipolu momenti, vibrācijas frekvences, reaktivitāte vai citi spektroskopiskie lielumi un šķērsgriezumi sadursmēm ar citām daļiņām.
J: Cik precīzas ir metodes, ko izmanto skaitļošanas ķīmijā?
A: Aprēķinu ķīmijā izmantoto metožu precizitāte ir no ļoti precīzas līdz ļoti aptuvenai. Ļoti precīzas metodes parasti ir iespējamas tikai mazām sistēmām.
J: Kā skaitļošanas ķīmija papildina eksperimentālos datus?
A: Parasti skaitļošanas ķīmija papildina informāciju, kas iegūta ar ķīmiskiem eksperimentiem. To var izmantot, lai prognozētu rezultātus, kas eksperimentāli vēl nav novēroti.
J: Vai pētāmās sistēmas lielums ietekmē to, cik daudz datoralaika ir nepieciešams?
A: Jā - palielinoties pētāmās sistēmas lielumam, pieaug arī analīzei nepieciešamais datorlaiks, kā arī tādi resursi kā atmiņas un diska vieta, kas nepieciešama glabāšanai.
Saistītie raksti
Autors
AlegsaOnline.com Datorķīmija: kas tā ir — metodes, īpašības un pielietojums Leandro Alegsa
URL: https://lv.alegsaonline.com/art/22297