Osteogenesis imperfecta (trauslo kaulu slimība) — cēloņi un simptomi
Osteogenesis imperfecta (trauslo kaulu slimība): cēloņi un simptomi — lūzumu risks, dzirdes zudums, diagnoze un atbalsts pacientiem. Uzziniet svarīgāko tagad.
Osteogenesis imperfecta ir ģenētiska slimība. To parasti sauc par trauslo kaulu slimību. Tā visbiežāk ir autosomāli dominējoša slimība, kas nozīmē, ka cilvēks var saslimt, ja tikai vienam no viņa vecākiem ir patoloģisks gēns. Tomēr ir arī retākas recesīvas formas. OI ietekmē kaulu sastāvdaļu, kas saistīta ar kolagēna struktūru — kolagēna “stieni”, kas nodrošina kaulu izturību. Gēnu izmaiņu rezultātā kolagēns kļūst vājāks vai trūkst, un kauli vieglāk lūst. Slimību 19. gadsimtā aprakstīja W. Vrolik, un kopš tā laika zināšanas par tās cēloņiem un ārstēšanu ir būtiski attīstījušās.
Attēlu galerija
10 Attēli







Simptomi un klīniskā aina
Osteogenesis imperfecta klīniskā aina ir ļoti mainīga — no viegliem gadījumiem ar dažiem lūzumiem līdz smagām formām jau dzimšanā. Galvenie simptomi:
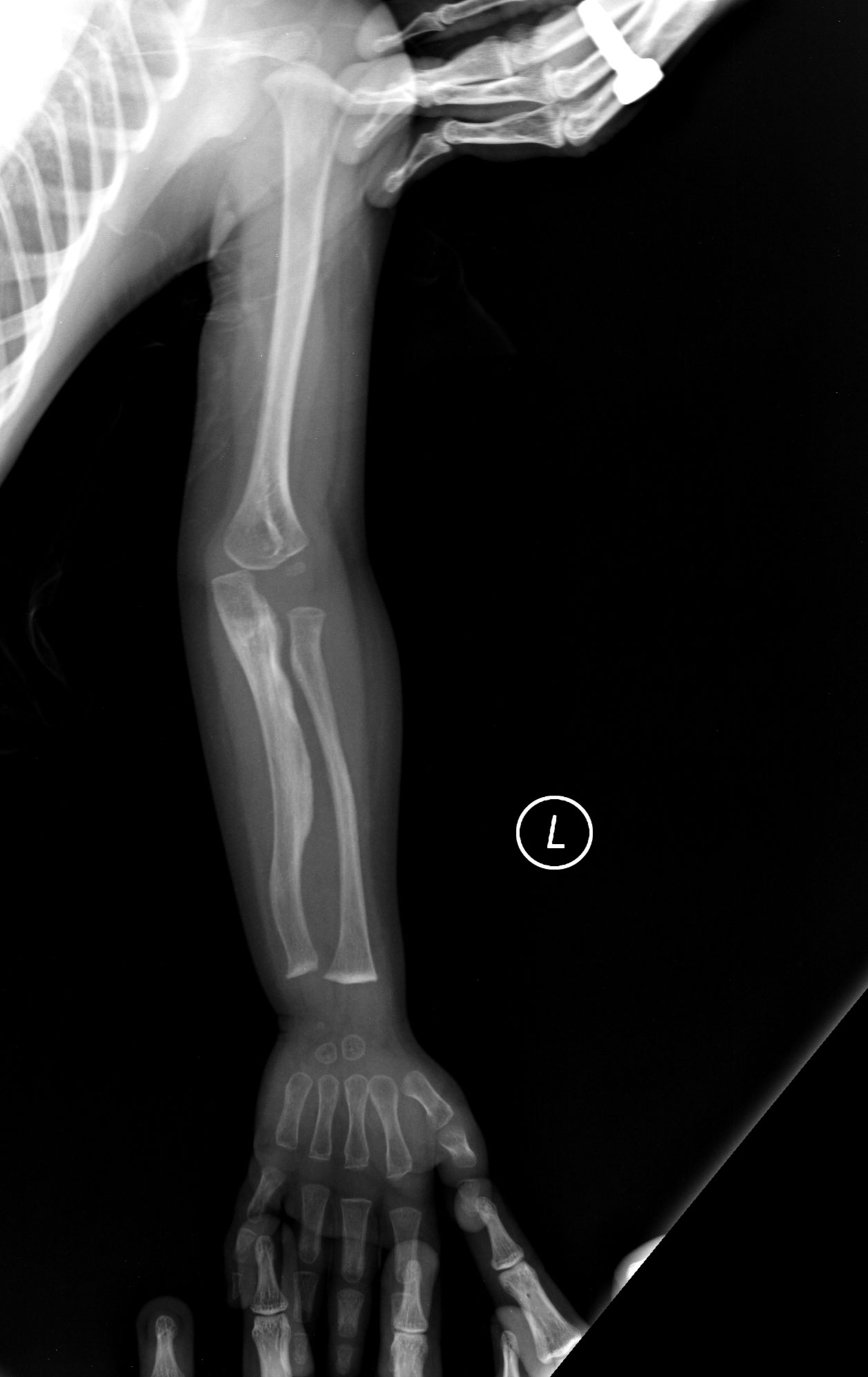
- Fraktūras: kaulu lūzumi bieži rodas pie mazākas traumas vai pat bez acīmredzama iemesla.
- Īss augums un skeleta deformācijas: deformācijas, liekumi, skolioze vai citi augšanas traucējumi.
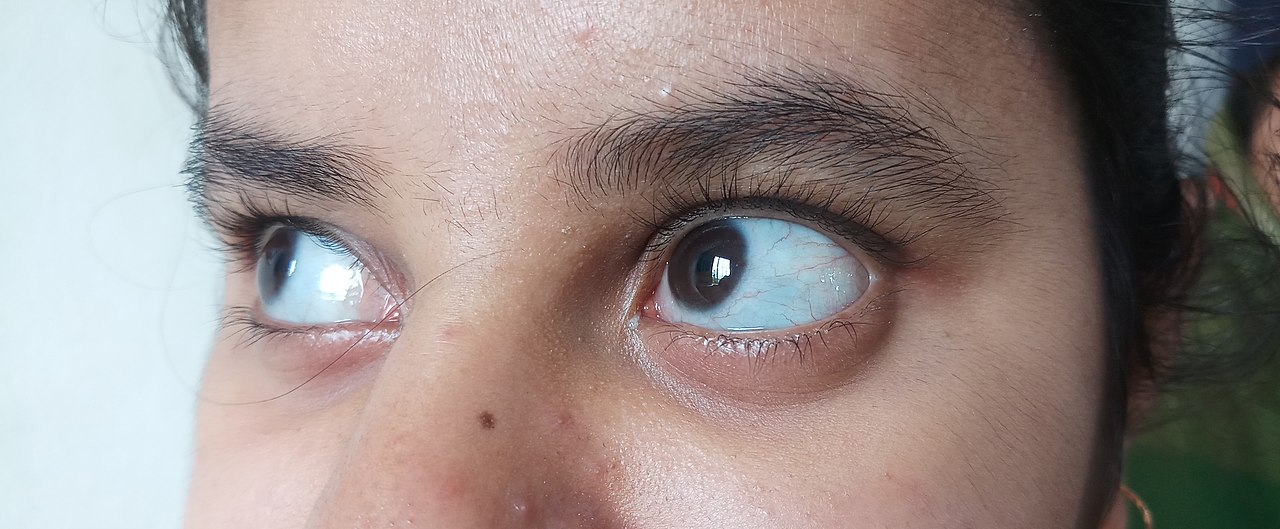
- Zilu vai violets acu sklera: sklera var izskatīties zilgana, jo caur to labāk redzams tajā esošais asinsvadu tīklojums.
- Dentu bojājumi (dentinogenesis imperfecta): trausli, mainīgas krāsas vai priekšlaicīgi nolietoti zobi.
- Locītavu laxity (saišu vājums): palielināta locītavu kustīgums.
- Dzirdes zudums: vidēji līdz smagi — aptuveni līdz pusei pieaugušo ar OI var attīstīties dzirdes pasliktināšanās.
- Elpošanas un sirds problēmas: smagākos gadījumos var būt plaušu hipoplāzija, repetitīvas plaušu infekcijas vai sirds un asinsvadu komplikācijas.
Slimības tipi
Tradicionāli OI iedala vairākos tipos (Sillence klasifikācija I–IV), kas atšķiras pēc smaguma un simptomiem:
- Tips I: visbiežāk sastopamais, salīdzinoši vieglāks, bieži ir normāla vai tuva normālai dzīves ilgums; var būt trausli zobi un atkārtotas fraktūras.
- Tips II: ļoti smags, bieži letāls jau perinatālā periodā; smagas kaulu deformācijas un daudz lūzumu.
- Tips III: progresējoši smags; daudzi lūzumi jau bērnībā, izteiktas skeleta deformācijas, var būt īss augums un dzirdes traucējumi.
- Tips IV: vidēji smags; vairāk simptomu nekā tipā I, bet parasti mazāk nekā tipā III.
Ir arī citas atšķirības un papildu tipi (V–VIII u. c.), kurām ir specifiski klīniskie un ģenētiskie īpašības.
Diagnoze
- Kliniskā novērtēšana: simptomi, ģimenes vēsture un fiziskā izmeklēšana (piem., zilas skleras, zobi, skeleta deformācijas).
- Radioloģija: rentgenogrammas, lai konstatētu fraktūras, kaulu struktūras izmaiņas un deformācijas.
- Kaulu blīvuma mērījumi (DXA): var palīdzēt novērtēt kaulu minerālu blīvumu.
- Ģenētiskā testēšana: analīze no asins parauga vai citiem audiem, lai noteiktu izmaiņas gēnos, kas saistīti ar kolagēnu (piem., COL1A1, COL1A2) vai citās saistītās ģenēs.
Ārstēšana un aprūpe
Osteogenesis imperfecta pilnībā nav izārstējama, taču mērķtiecīga ārstēšana un interdisciplināra aprūpe var samazināt fraktūru skaitu, uzlabot funkciju un dzīves kvalitāti. Galvenās pieejas:
- Medikamentoza terapija: bisfosfonāti (piem., pamatojoties uz pediatrijas vai pieaugušo ieteikumiem) samazina kaulu resorbciju un var palielināt kaulu blīvumu un samazināt lūzumu risku.
- Ortopēdiska ķirurģija: intramedulāri stieņi (rodding) un citas operācijas var stabilizēt izliektus vai bieži lūstošus kaulus, uzlabot funkcionalitāti un samazināt atkārtotu fraktūru risku.
- Fizioterapija un rehabilitācija: muskuļu stiprināšana, staigāšanas un kustību prasmju uzturēšana, drošu kustību iemaņu apmācība, ortozes un palīglīdzekļu izmantošana.
- Sāpju vadība: medikamentoza un nemedikamentoza pieeja, fizioterapija un psiholoģiska atbalsta metodes.
- Stomatoloģiska aprūpe: regulāra zobārsta uzraudzība, ārstēšana dentinogenesis imperfecta gadījumos.
- Audioloģiskā uzraudzība: regulāras dzirdes pārbaudes un nepieciešamības gadījumā dzirdes aparāti vai citas ierīces.
- Dzīvesveida ieteikumi: pietiekama kalcija un D vitamīna uzņemšana, droši fiziskie vingrinājumi, smēķēšanas izvairīšanās un profilaktiskas stratēģijas samazināt traumas risku.
Komplikācijas un prognoze
Prognoze ļoti atkarīga no slimības tipa un smaguma pakāpes. Vieglākos gadījumos cilvēki var dzīvot pilnvērtīgu dzīvi ar rūpīgu aprūpi; smagākos — var būt nopietnas kustību, elpošanas vai neiroloģiskas sekas. Komplikācijas var ietvert hronisku sāpi, mobilitātes ierobežojumus, atkārtotas plaušu infekcijas un dzirdes zudumu. Agrīna diagnostika, atbilstoša ārstēšana un daudznozaru komanda (ortopēds, ģenētiķis, fizioterapeits, zobārsts, audiologs u. c.) uzlabo iznākumu.
Ģenētiskā konsultācija
Ja ģimenē ir OI vai ir aizdomas par to, ieteicama ģenētiskā konsultācija. Konsultācija palīdz saprast mantojuma riskus, pieejamās ģenētiskās pārbaudes (ieskaitot preimplantācijas diagnostiku un prenatālas iespējas) un ģimenes plānošanas iespējas.
Praktiski padomi ikdienai: pielāgot mājas vidi drošai pārvietošanai, izmantot aizsarglīdzekļus bērniem, plānot regulāras medicīniskās pārbaudes un sadarboties ar rehabilitācijas speciālistiem, lai uzturētu kustību spējas un samazinātu lūzumu skaitu.
Simptomi
Mazāk smagi OI simptomi var būt:
- viegli lauzti kauli
- vaļīgi savienojumi
- zems muskuļu tonuss
- zila, violeta vai pelēka parasti baltās acu daļas krāsa.
- trīsstūrveida sejas forma
- tendence saslimt ar skoliozi
- trausli zobi
OI ir arī daudzi citi nopietni un letāli simptomi, tostarp elpošanas traucējumi un kaulu deformācija.
Demogrāfiskie dati
Jautājumi un atbildes
J: Kas ir osteogenesis imperfecta?
A: Osteogenesis imperfecta ir ģenētiska slimība, ko parasti sauc par trauslo kaulu slimību. Tā novājina vai iznīcina kolagēna stieņus, kas nodrošina kaulu stiprību, un tās rezultātā kauli biežāk lūst.
J: Kā tiek pārmantota osteogenesis imperfecta?
A: Osteogenesis imperfecta parasti ir autosomāli dominējoša slimība, kas nozīmē, ka cilvēks var saslimt ar šo slimību, ja tikai vienam no viņa vecākiem ir patoloģisks gēns.
J: Kas pirmais identificēja osteogenesis imperfecta?
A: Vroliks pirmais 1849. gadā identificēja osteogenesis imperfecta.
Vai osteogenesis imperfecta ir ārstējama?
A: Diemžēl osteogenesis imperfecta nav ārstējama.
J: Kādi ir četri osteogenesis imperfecta veidi?
A: Četri osteogenesis imperfecta veidi ir pirmais, otrais, trešais un ceturtais tips.
J: Kādi ir trešā tipa osteogenesis imperfecta simptomi?
A: Cilvēkiem ar trešā tipa osteogenesis imperfecta var būt vairāk nekā 100 lūzumu pirms pubertātes. Viņu acis bieži iegūst violetu, zilu vai pelēku nokrāsu, un cilvēkiem ar šo tipu bieži ir arī dzirdes zudums.
J: Cik bieži cilvēkiem ar osteogenesis imperfecta ir dzirdes zudums?
A: 50 % cilvēku ar osteogenesis imperfecta ir dzirdes zudums, kļūstot pieaugušiem.
Saistītie raksti
Autors
AlegsaOnline.com Osteogenesis imperfecta (trauslo kaulu slimība) — cēloņi un simptomi Leandro Alegsa
URL: https://lv.alegsaonline.com/art/73422
Avoti
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"