Molekulārās orbītas (MO): definīcija, nozīme un elektroniskā uzbūve
Uzzini molekulāro orbitāļu (MO) definīciju, nozīmi un elektronisko uzbūvi — skaidrojums, diagrammas un pielietojums kvantu ķīmijā saprotamā valodā.
Ķīmijā ar molekulāro orbitāli (jeb MO) skaidro, kas notiek ar elektroniem, kad atomi savienojas molekulā. MO ir matemātiska funkcija, kas apraksta viļņveida elektronu uzvedību molekulā — tās kvadrāts dod varbūtību atrast elektronu konkrētā apgabalā. Ķīmiķi izmanto šādas funkcijas, lai prognozētu vai izskaidrotu ķīmiskās un fizikālās īpašības, piemēram, saistību spēku, magnētiskumu, elektroniskās pārejas un reaktivitāti. Ar MO palīdzību var precīzāk noteikt varbūtību, ka elektrons atradīsies kādā konkrētā apgabalā molekulā, un tāpēc šis rīks ir centrāls, lietojot kvantu mehāniku molekulu skaidrošanā.
Ķīmijā MO parasti modelē, apvienojot atsevišķu atomu orbitāles vai to hibrīdus. Ķīmiķi bieži izveido molekulāro orbitāļu matemātiskos modeļus, izmantojot atomu orbitāles kā bāzes funkcijas (LCAO — linear combination of atomic orbitals). Var izmantot arī hibrīdorbitāles no katra molekulas atoma vai citus MO risinājumus, ko aprēķina ar datoriem. Ar šīm funkcijām var strādāt, lai saprastu, kā un kāpēc atomi molekulās turas kopā, kā arī lai prognozētu eksperimentālas īpašības.
Attēlu galerija
5 Attēli
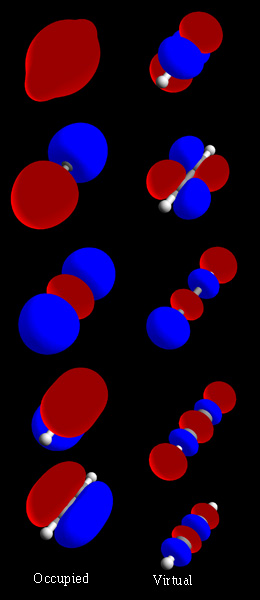



Kā veidojas molekulārās orbītas
Molekulārā orbitāle rodas, kad atomu orbitāles pārklājas un interferē. Šajā procesā divas pamatprincipālas kombinācijas ir iespējamas:
- Konstruktīvā interferenca — atoma orbitāļu fāzes sakrīt, kas rada saistošu (bonding) orbitāli ar zemāku enerģiju un lielāku elektronu blīvumu starp atomu kodoliem.
- Destruktīvā interferenca — fāzes neatbilst, radot antisaistošu (antibonding) orbitāli ar pogu enerģiju un urbumu (nodes) starp atomu kodoliem.
Jo vairāk kodolu attālums un orbitāļu pārklājums, jo nozīmīgāka ir šo kombināciju ietekme uz MO enerģiju un formu. Katram molekulāram sistēmai kvantmehāniski risina viļņfunkciju vienādojumu (apvienojot elementus, piemēram, ar Hartree–Fock vai DFT metodēm), lai noteiktu konkrētās MO enerģijas un formas.
Veidi un īpašības
Galvenie orbītu tipi molekulās ir:
- σ (sigma) orbītas — simetriskas ap saites asi, rodas no s vai p_z orbitāļu galvenā pārklāšanās. Tām parasti nav nodu starp kodoliem.
- π (pi) orbītas — veidojas no p orbitāļu sānu pārklāšanās; tām ir nodu plakne, kas iet caur saites asi.
- δ, φ u. c. — sarežģītākas simetrijas orbitāles var parādīties pārejas metālu kompleksos un lielās sistēmās.
Antisaistošajām orbitālēm bieži piemīt vairāk nodu (viļņu nulles), un to enerģija ir augstāka. Kopējā orbītāļu enerģijas kārtība nosaka, kur elektroni izvietosies saskaņā ar Paulija izslēgšanas principu un Hunda noteikumiem.
HOMO, LUMO un saites spēks
HOMO (Highest Occupied Molecular Orbital) ir augstākā aizpildītā orbitāle, bet LUMO (Lowest Unoccupied Molecular Orbital) — zemākā neaizpildītā. Starpība HOMO–LUMO bieži korelē ar molekulas ķīmisko reaktivitāti un optiskajām īpašībām: mazāka starpība parasti nozīmē vieglāku elektroniskās pārejas excitation un lielāku reaktivitāti.
Vienkārša saites kārtības (bond order) aprēķina formula ir:
Saite = (nbonding − nantibonding) / 2
Piemēri: H2 — viena saite (bond order = 1); He2 — bond order = 0 (neeksistē stabila saite); O2 — divi nepāra elektroni HOMO dēļ, kas padara to par paramagnētisku.
Delokalizācija, konjugācija un aromātiskums
Molekulārās orbītas var būt lokālas (ierobežotas pie viena atomu pāra) vai delokalizētas (izstieptas pāri vairākiem atomiem). Delokalizācija ir pamats konjugētām sistēmām un aromātiskumam: viengabalaina π orbītu sistēma ļauj elektroniem brīvāk kustēties pa molekulu, kas ietekmē stabilitāti, krāsu un elektrisko vadītspēju.
Praktiskā nozīme un kalkulācijas
Ķīmiķi un materiālzinātnieki izmanto MO teoriju, lai skaidrotu un prognozētu:
- ķīmisko reaktivitāti un reakciju ceļus, izmantojot HOMO/LUMO saskaņojumu;
- magnētiskās īpašības (piem., paramagnētisms O2);
- elektroniskās spektroskopijas pārejas (UV/Vis), jo MO sadalījums nosaka iespējamo pāreju enerģijas;
- molekulāro dizainu — piemēram, organisko pusvadītāju, katalizatoru un zāļu dizainā.
Praktiski MO aprēķini tiek veikti ar dažādām kvantķīmijas metodēm — LCAO-MO pieeja, Hartree–Fock, post-Hartree–Fock metodes un Density Functional Theory (DFT). Datoru programmas ļauj vizualizēt orbītāļu formu un blīvumu, kas palīdz interpretēt eksperimentālos datus.
Limitācijas un piezīmes
Kaut arī MO teorija ir ļoti noderīga, tai ir ierobežojumi — precizitāte atkarīga no izmantotā bāzes kopuma un aprēķina metodes. Dažos gadījumos, īpaši sistēmās ar spēcīgām korrelācijām (piem., pārejas metālu kompleksos), ir nepieciešamas sarežģītākas metodes, lai iegūtu pareizu enerģiju un elektronu sadalījumu.
Orbitāļu diagrammas un MO modeļi nodrošina spēcīgu intuīciju par to, kā elektroni organizējas molekulās, un ir centrālais instruments mūsdienu ķīmijā — no pamatprincipiem līdz praktiskai materiālu un zāļu izstrādei.

Vēsture
Vārdu "orbitālais" angļu valodā pirmo reizi lietoja Roberts S. Mullikens. Vācu fiziķis Ervins Šrēdingers par MO rakstīja jau agrāk. Šrēdingers tās sauca par Eigenfunktion.
Fiziķis Makss Borns 1926. gadā aprakstīja molekulāro orbitāļu teoriju. Mūsdienās tā ir pazīstama kā Borna likums un ir daļa no kvantu mehānikas Kopenhāgenas interpretācijas. Sākotnēji, kad šī teorija tika ierosināta, tā nesaskanēja ar Nils Bora atoma modeli. Bora modelī elektroni bija aprakstīti kā "riņķojoši" ap kodolu, pārvietojoties pa apli. Tomēr Borna modelis galu galā guva plašu atbalstu, jo tas spēja aprakstīt elektronu atrašanās vietas molekulās un izskaidroja vairākas iepriekš neizskaidrojamas ķīmiskās reakcijas.
Pārskats
Atoma orbitāles paredz elektronu atrašanās vietu atomā. Molekulārās orbitāles veidojas, kad atomu orbitāles tiek apvienotas. Molekulārā orbitāle var sniegt informāciju par molekulas elektronu konfigurāciju. Elektronu konfigurācija ir viena(-u) elektrona(-u) visticamākā atrašanās vieta un enerģija. Visbiežāk MO attēlo kā atomu orbitāļu lineāru kombināciju (LCAO-MO metode), īpaši aptuvenā lietojumā. Tas nozīmē, ka ķīmiķi pieņem, ka iespēja, ka elektrons atradīsies jebkurā molekulas punktā, ir elektronu atrašanās varbūtību summa, pamatojoties uz atsevišķām atomu orbitālēm. LCAO-MO ir vienkāršs saišu modelis molekulās, un tas ir svarīgs molekulāro orbitāļu teorijas izpētē.
Ķīmiķi teorētiķi izmanto datorus, lai aprēķinātu dažādu molekulu MO (gan reālo, gan iedomāto). Dators var uzzīmēt "mākoņa" grafikus, lai parādītu, cik liela ir varbūtība, ka elektrons atradīsies jebkurā reģionā. Datori var arī sniegt informāciju par molekulas fizikālajām īpašībām. Tie var arī pateikt, cik daudz enerģijas ir nepieciešams, lai veidotu molekulu. Tas palīdz ķīmiķiem pateikt, vai dažas mazas molekulas var apvienot, lai izveidotu lielākas molekulas.
Lielākā daļa mūsdienu aprēķinu ķīmijas metožu sākas ar sistēmas MO aprēķināšanu. Katra MO elektrisko lauku rada visu atomu kodoli un vidējais pārējo elektronu sadalījums.
Analoģija
Izpratne par MO ir līdzīga uzdevumam, kas ir līdzīgs uzdevumam uzzināt, kur atrodas katrs darbinieks lielā mājokļa labiekārtošanas veikalā (neapskatot veikala iekšpusi). Analītiķis zina veikalā strādājošo darbinieku skaitu un katra darbinieka nodaļu. Viņš arī zina, ka darbinieki nestājas viens otram uz kājām un darbinieki stāv ejā, nevis preču plauktos. Darbinieki atstāj savu nodaļu, lai palīdzētu klientiem atrast preces citās nodaļās vai pārbaudītu inventāru. Analītiķis, kas nosauc visu veikalā esošo darbinieku atrašanās vietu izvēlētajā brīdī, neieskatoties iekšā, ir kā ķīmiķis, kas aprēķina molekulas MO. Tāpat kā MO nevar noteikt katra elektrona precīzu atrašanās vietu, nav zināma katra darbinieka precīza atrašanās vieta. MO, kam ir mezgla plakne, ir kā secinājums, ka darbinieki staigā pa ejām, nevis pa plauktiem. Lai gan elektroni tiek ieguldīti no konkrēta atoma, elektrons aizpilda MO, neņemot vērā tā avota atomu. Tas ir tāpat kā darbinieks, kurš dienas laikā atstāj savu nodaļu, lai staigātu citur veikalā. Tātad MO ir nepilnīgs elektronu apraksts, tāpat kā analītiķa aprēķini par neredzamo veikalu ir nepilnīgs minējums par darbinieku atrašanās vietām.

Molekulāro orbitāļu veidošanās
Ķīmiķi teorētiķi ir izgudrojuši noteikumus MO aprēķināšanai. Šo noteikumu pamatā ir izpratne par kvantu mehāniku. Kvantu mehānika palīdz ķīmiķiem izmantot fizikas atziņas par elektroniem, lai noskaidrotu, kā elektroni uzvedas molekulās. Molekulārās orbitāles veidojas no "atļautās" mijiedarbības starp atomu orbitālēm. (Mijiedarbība ir "atļauta", ja atomu orbitāļu simetrijas (ko nosaka grupu teorija) ir savstarpēji saderīgas.) Ķīmiķi pēta atomu orbitāļu mijiedarbību. Šīs mijiedarbības rodas no divu atomu orbitāļu savstarpējās pārklāšanās (mēraukla tam, cik labi divas orbitāles savstarpēji konstruktīvi mijiedarbojas). Pārklāšanās ir svarīga, ja atomu orbitāļu enerģijas ir tuvas. Visbeidzot, MO skaitam molekulā ir jābūt vienādam ar atomu orbitāļu skaitu atomos, kas tiek apvienoti, lai veidotu molekulu.
Kvalitatīvā pieeja
Ķīmiķiem ir jāsaprot MO ģeometrija, lai apspriestu molekulu struktūru. LCMO (lineārā atomu orbitāļu molekulāro orbitāļu kombinācija) metode sniedz aptuvenu, bet labu MO aprakstu. Izmantojot šo metodi, molekulārās orbitāles izsaka kā visu molekulas atomu atomu orbitāļu lineārās kombinācijas.
Atomu orbitāļu lineārās kombinācijas (LCAO)
Molekulārās orbitāles pirmo reizi 1927. un 1928. gadā ieviesa Frīdrihs Hunds un Roberts S. Mullikens.
Atomu orbitāļu lineāro kombināciju jeb "LCAO" aproksimāciju molekulārām orbitālijām 1929. gadā ieviesa sers Džons Lennards-Džonss. Viņa revolucionārais darbs parādīja, kā no kvantu principiem iegūt fluora un skābekļa molekulu elektronisko struktūru. Šī kvalitatīvā pieeja molekulāro orbitāļu teorijai ir daļa no mūsdienu kvantu ķīmijas aizsākumiem.
Atomu orbitāļu lineārās kombinācijas (LCAO) var izmantot, lai uzminētu molekulārās orbitāles, kas veidojas, kad molekulas atomi savienojas. Līdzīgi kā atoma orbitālei, arī molekulārajai orbitālei var sastādīt Šrēdingera vienādojumu, kas apraksta elektrona uzvedību. Atomu orbitāļu lineārās kombinācijas (atomu viļņfunkciju summas un starpības) nodrošina aptuvenus risinājumus molekulārajiem Šrēdingera vienādojumiem. Vienkāršām divatomu molekulām iegūtās viļņu funkcijas matemātiski attēlo vienādojumi
Ψ = c ψ aa+ c ψ bb
un
Ψ* = c ψ aa- c ψ bb
kur Ψ un Ψ* ir attiecīgi saistošo un anti-saistošo molekulāro orbitāļu molekulārās viļņu funkcijas, ψa un ψb ir attiecīgi atomu a un b atomu viļņu funkcijas, bet ca un c bir regulējamie koeficienti. Šie koeficienti var būt pozitīvi vai negatīvi atkarībā no atsevišķu atomu orbitāļu enerģijām un simetrijas. Diviem atomiem kļūstot tuvāk vienam otram, to atomu orbitāles pārklājas, radot augsta elektronu blīvuma apgabalus. Tādējādi starp abiem atomiem veidojas molekulārās orbitāles. Atomus kopā satur elektrostatiskā pievilkšanās starp pozitīvi lādētiem kodoliem un negatīvi lādētiem elektroniem, kas aizņem saistošās molekulārās orbitāles.
Saistošie, antisaistošie un nesaistošie MO
Kad atomu orbitāles mijiedarbojas, var rasties trīs veidu molekulārās orbitāles: saistošā, anti-saistošā vai nesaistošā.
Saistošie MO:
- Saišu mijiedarbība starp atomu orbitālēm ir konstruktīva (fāzveida) mijiedarbība.
- Saistošo MO enerģija ir zemāka nekā atomu orbitāļu enerģija, kas apvienojas, lai tās radītu.
Pretsavienojošie MO:
- Antisavienojošās mijiedarbības starp atomu orbitālēm ir destruktīvas (ārpusfāžu) mijiedarbības.
- Anti-savienojošo MO enerģija ir augstāka nekā atomu orbitāļu enerģija, kas apvienojas, lai tās radītu.
Nesaistītie MO:
- Nesavienojošie MO ir rezultāts tam, ka nav mijiedarbības starp atomu orbitālēm, jo nav saderīgu simetriju.
- Nesaistītajām MO būs tāda pati enerģija kā viena molekulas atoma atoma orbitālei.
HOMO un LUMO
Katrai molekulārajai orbitālei ir savs enerģijas līmenis. Ķīmiķi šķiro MO pēc enerģijas līmeņiem. Ķīmiķi pieņem, ka elektroni vispirms aizpildīs zemākā enerģijas līmeņa MO. Piemēram, ja molekulā ir elektroni, ar kuriem aizpildīt 15 orbitāles, tad tiks aizpildītas 15 MO ar zemāko enerģijas līmeni. Saraksta 15. MO sauc par "visaugstāk aizņemto molekulāro orbitāliju" (HOMO), bet saraksta 16. MO ir "zemākā neaizņemtā molekulārā orbitāle" (LUMO). Starpību starp HOMO enerģijas līmeni un LUMO enerģijas līmeni sauc par joslas spraugu. Joslu spraugu dažkārt var izmantot kā molekulas uzbudināmības mērauklu: jo mazāka ir tās enerģija, jo vieglāk to uzbudināt. Kad elektrons ir uzbudināts, tas pāriet uz neaizņemtu MO. Piemēram, tas var palīdzēt noteikt, vai kaut kas izstaros gaismu (luminiscence).

Jautājumi un atbildes
J: Kas ir molekulārā orbitāle?
A: Molekulārā orbitāle (jeb MO) ir matemātiska funkcija, kas apraksta viļņveida elektronu uzvedību molekulā. Tā izskaidro, kas notiek ar elektroniem, kad atomi savienojas molekulā, un var noteikt varbūtību, ka elektronu var atrast kādā konkrētā apgabalā.
J: Kā ķīmiķi veido molekulāro orbitāļu matemātiskos modeļus?
A: Ķīmiķi parasti veido molekulāro orbitāļu matemātiskos modeļus, apvienojot atomu orbitāles. Var izmantot arī hibrīdorbitāles no katra molekulas atoma vai citas molekulārās orbitāles no atomu grupām. Ar šīm funkcijām var strādāt datori.
J: Kāda saistība kvantu mehānikai ir ar molekulu pētīšanu?
A: Molekulārās orbītas ļauj ķīmiķiem izmantot kvantu mehāniku molekulu pētīšanā. Tās atbild uz jautājumiem par to, kā atomi molekulās turas kopā, un sniedz ieskatu ķīmiskajās un fizikālajās īpašībās.
J: Kas ir orbitāļu diagrammas?
A: Orbitāļu diagrammas ir vizuāli attēli, kas norāda, kur atomā visdrīzāk varētu atrasties elektroni, pamatojoties uz to dažādajām noapaļotajām formām.
J: Kā darbojas hibrīda orbitāles?
A: Hibrīdās orbītas apvieno dažādu veidu atomu orbītas vienā jaunā veidā, kam ir unikālas īpašības salīdzinājumā ar tā sastāvdaļām. Šos hibrīdus bieži izmanto, veidojot molekulāro orbitāļu matemātiskos modeļus.
J: Kā datori var palīdzēt izpētīt MO?
A: Datori var palīdzēt MO pētīšanā, strādājot pie to funkcijām un sniedzot precīzākus ķīmisko un fizikālo īpašību paredzējumus vai skaidrojumus molekulās.
Saistītie raksti
Autors
AlegsaOnline.com Molekulārās orbītas (MO): definīcija, nozīme un elektroniskā uzbūve Leandro Alegsa
URL: https://lv.alegsaonline.com/art/65861
Avoti
- nobelprize.org : Nobelprize.org